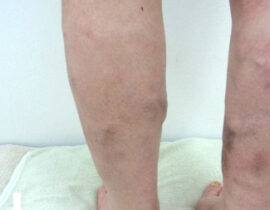
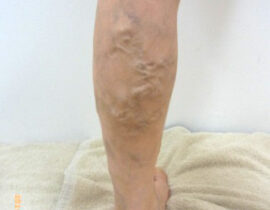
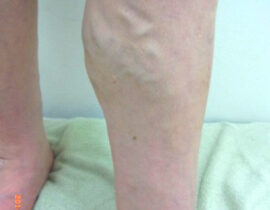
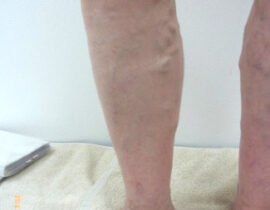
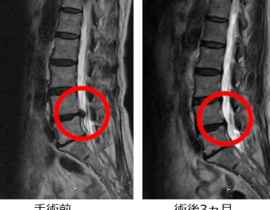
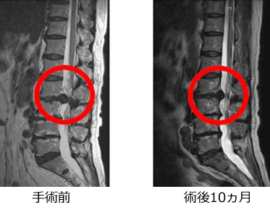
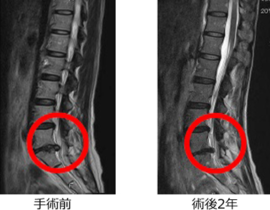
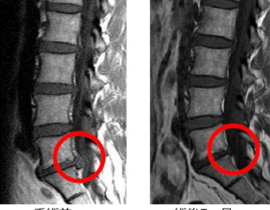
薬剤発案者へのQ&A
「CDC6shRNA治療」の薬剤の開発者に、がん遺伝子治療について6つの質問をしました。和訳と原文をご紹介します。
質問・回答の和訳
質問1.
本治療を要約すると「組み換えRNAにCDC6 RNAi とhTERT(ヒトテロメラーゼ逆転写酵素)を搭載して、がん細胞に送達し、mRNAi(RNA干渉)によりがん細胞中のCDC6を消去して、がんの無限増殖を止め、がん細胞の老化ないしはアポトーシス(自殺)を誘導する療法」ということになると思いますが、端的かつ適切に表現できないでしょうか。「遺伝子治療」「ウイルス療法」「RNA療法」「mRNAiがん療法」・・・など適切な評伝はないでしょうか。
回答1.
私は一種の遺伝子治療と考えています。とりわけ「無侵襲のがん根絶療法」といえます。
質問2.
CDC6タンパクを消去しても治療効果が得られないがんはありませんか。
回答2.
私は医師ではないので、どのがんがCDC6 RNAi 療法に反応しないかテストすることができません。ただ、がんにはかなりの種類があり同じがんの中にも複数のタイプがありますので、中にはCDC6RNAi療法が効きにくいものがあるかもしれません。私が行った細胞レベルでの検査では、ヒトの神経芽細胞種の複数のタイプの中で、KANRというタイプのものはCDC6RNAi療法に反応しませんでした。このKANRの他に別のタイプの13種類の神経芽細胞種を対象としてその中にCDC6タンパクが発現しているかどうか調べたところ、KANRと別の2種類のタイプにはCDC6タンパクが確認されませんでした。すなわち、これらはCDC6 RNAi 療法のターゲットが無いので、本療法では腫瘍の発育を抑制することはできないことになります。ただし、これらCDC6タンパクが発現していないタイプのものは細胞培地で殆ど発育しませんでした。 ※そもそもCDC6タンパクが発現していなければそれががんであっても殆ど発育しない。
質問3.
女性器などにはCDC6タンパクの発現が見られるようだが、仮に健常人に本治療が行われた場合その安全性は担保されるのでしょうか。
回答3.
これは非常に良い質問です。私たちの治療試薬は、正常細胞であれがん細胞であれ、全ての細胞の中に侵入していきますが、CDC6 RNAi は非常に特殊な振る舞いをします。CDC6 RNAi がその作用を開始するためには、hTERT(ヒトテロメラーゼ逆転写酵素)プロモーターにより作動スイッチが入れられなければいけません。この転写コントロールを行う特殊な遺伝子要素は、hTERTゲノム中のエクソン1の部位にあるEBOXです。正常細胞において、いつ転写を開始、いつ終えるかの指示を出すのがこのEBOXです。この調整機能は殆ど全ての正常細胞で維持されており、hTERTプロモーターの活動は停止しています。しかし、殆どの人のがん細胞ではゲノムが不安定な状態になっているため、この制御機能が失われてhTERTプロモーターの活動が開始して転写が開始されてしまいます。この特殊な作用を持つEBOXにより正常細胞ではCDC6 RNAi の発現が抑えられて転写がおきないように制御されています。がん細胞においてのみ、このEBOXの制御が失われてhTERTの活動性が高まりCDC6 RNAi の転写が活発に行われるのです。
質問4.
点滴やカテーテル治療により本薬剤はがん細胞に有効に送達されますか。血液中で失活しませんか。また、がん性腹膜炎や胸膜炎において、腹腔内や胸腔内へ本製剤を直接注入しても、腫瘍表面から薬剤が細胞内へ送達されますか。すなわち本製剤をがん細胞へ直接注入する以外に有効な送達法はありますか。
回答4.
治療薬の構成物は、ヒトの細胞から培養された組織が派生したものなので、血管へ注射により体内に送り込むことはできますが、短時間で失活してしまいます(血中では2時間未満で失活します)。このため腫瘍(がん)に直接注射するのが最善の方法です。しかし、実際にはそれが難しい場合もあります。その欠点を回避するために、私たちの研究所では両親媒性の微粒子を開発しました。これはCDC6 RNAi を運んで機能させるプラスミドでcytonacxと命名しました。この新しいタイプの治療薬は血液中で安定しており、大きな分子複合体で生体内で他の成分と共存でき、かつ生体内で分解されるので、静脈注射で生体内へ送り込むことができます。
質問5.
本治療薬を腫瘍に直接注入するとして、腫瘍のサイズ、量に応じて、それを完全に除去するために必要な薬剤量を理論的に算出することはできますか。例えば直径1㎝のがん細胞を完全に除去するのに総量で何mlあれば良いのですか。
回答5.
算出できません。必要量を決定するためのヒントとなる文件を探してみます。 ※この質問をした当時は至適投与量算出の情報はありませんでしたが、 その後の治療経験からがんの広がりや病期に応じて必要投与量を決定しています。 末期がんに対する治療が中心になっているため何ml投与すれば完全に治せるというものではありません。がんの病勢をコントロールした後、定期的に追加投与をすることでがんを眠らせ続ける(自殺させる)ことを目指します。
質問6.
がん種の中で本治療が最も有効と考えられるのは?
回答6.
私たちが当初本薬剤を設計したときは、がん細胞がそのDNA複製の開始点でCDC6タンパクを利用する限り、本薬剤はがん細胞の複製を抑えると考えられました。しかし、例えば成長が遅く分裂が乏しい胃がんのように、多くのがんに対してはCDC6 RNAi 治療薬の効果の評価は難しいのです。私が思うに、この種のがんに対して本治療薬のみで治療効果を上げるのは難しいです。しかし、特にp53 p16 pGAdd45a を搭載したcytonacxを利用すれば、良い治療効果が得られると思います。 本治療薬の創始者であるLuo Feng博士からこのように直接回答を得られたことで、「有効な治療法はないと宣告されながらも、何か治癒につながる治療法はないかと模索している多くのがん患者さんたちに、この治療を実際に提供する気になったともいえる。
薬剤発案者への質疑応答(原文)
question 1
If I understand correctly, this therapy in general terms seems to attach and hTERT to recombinant RNA in order to be delivered to the cancer cell and then knocks down the CDC6 in the cancer cell through mRNAi to arrest the unlimited proliferation of the cancer and cause cellular senescence and/or apotosis. I was wondering if there may be a more succinct way of explaining this. Similarly, have you decided on an appropriate naming for this therapy? Such names as gene or genetic therapy, virus therapy, RNA therapy or mRNA therapy come to mind.
For your question 1:
I think this is a kind of gene therapy, in specific, it is a Non-Invasive Cancer Eradication (NICE) treatment;
question 2.
Have you found any cancers for which knocking down the CDC6 is not effective?
For your question 2:
I am not a doctor, so I could not have chance to test which kind of human cancer is resistant to CDC6 shRNA. I believe that there might be many kinds of cancers, or different types of same cancer that are resistant to CDC6 shRNA mediated treatment. In my cell tests, a human neuroblastoma cell line, KANR, displayed no response to CDC6 shRNA. We have examined the expression of Cdc6 in this tumor cells and other 13 different primary neuroblastoma cells. KANR, with other two cell lines could not be positive for Cdc6 expression. Therefore, CDC6 shRNA is hardly find its target in the tumor cells, doing nothing on suppression of tumor cell growth, though this kind of tumor cell grow poorly in the culture.
question 3.
The expression of CDC6 is also seen in, for instance, female genitalia. Is the safety of this therapy guaranteed even if used in healthy individuals?
For your question 3:
This is a very good question. I only can say our therapeutic reagents enter cells whether it is normal or cancerous one, but the production of CDC6 shRNA will be much different. The CDC6 shRNA expression is driven and controlled by human telomerase reverse transcriptase (hTERT) promoter. In this specific transcription control gene element, a novel E-box located in the exon 1 of the hTERT genome. It is this downstream E-box that can tell when the transcription should be on and when should be off in normal cells. This regulation mechanism is maintained in most normal somatic cells and keeps the hTERT promoter activity in repression status. However, as results of genome instability in many kinds of human cancers, this control function is lost in these cancer cells, leading to the activation of transcription. We use E-Box this specificity to control CDC6 shRNA expression and to let the transcription in normal cells or in cells like you have cited to be silent. Only in cancer cells in which hTERT activity is higher is the CDC6 shRNA transcription activated due to the E-Box repression effect has been lost.
question 4.
Can this therapy be effectively delivered to tumors intravenously orvia catheter, or does the therapy become deactivated in the bloodstream. Likewise, in the case of cancerous peritonitis or pleurisy can the drug be effectively delivered into the tumor cells from the tumor surface if injected into the peritoneal or thoracic cavities, respectively? In other words, is there no other treatment than to directly inject the drug into the tumor?
For your question 4:
Due to final forms of therapeutic components are derivatives of tissue cultured human cells, they can be introduced into body intravenously, but will be inactivated or degraded very soon (less than 2 hours in blood stream). For this reason, direct injection into tumor foci is best way. However, it is hard to accomplish in practice. To circumvent this drawback, our RNTein Biotech Lab. has developed an amphiphilic nanoparticle, we called Cytonacx carrying CDC6 shRNA producing plasmid. This novel kind of therapeutic reagent is stable in bloodstream and its large molecular co-polycomplex is biocompatible and biodegradable, and can be introduced into body intravenously.
question 5.
With this drug, assuming direct injection, is it theoretically possible to determine the volume required to completely eliminate a tumor based on its total mass. For instance are you able to determine how many ml are required to eliminate a tumor 1 cm in diameter?
For your question 5:
My answer is no. I will find some reference which may help you to do some estimation.
question 6.
Of the various carcinoma, is there one in particular, such as gastric carcinoma or poorly-differentiated carcinomas, for which you consider this therapy to be most effective?
For your question 6:
When we designed AJS2001, it will suppress cancer cell proliferation as long as the cell makes use of Cdc6 in the initiation of DNA replication. However, many tumors, such as human gastric tumor with slow growth and poor differentiation, the effect of CDC6 shRNA mediated treatment is hard to evaluate. I really think that AJS2001 alone would do poor job in treating such kind of tumor. However, combining with Cytonacx, specifically use Cytonacx carrying some tumor suppression protein-producing vector, such as p53, p16, pGAdd45a etc. may produce nice results.
その他の質疑応答
Dear Dr. Luo.Feng Although I know you are quite busy, could I bother you with a few questions I had while reviewing your articles and papers. 1. In the paper on AJS2001, you touch briefly on the Vpr protein. Is it currently contained in the therapy agent or is CDC6 shRNA loaded in its place. 2. If Vpr is not contained in the agent we are currently using, was Vpr contained in the medicine used in the Japanese case of scirrhous gastric cancer treated in 2006? I would appreciate any enlightenment you can give me on this. 3. If it is the case that Vpr is no longer used, could you explain why it is no longer used. 4. How many vectors (recombinant RNA) are there in each milliliter of AJS2001 and Cytonax? 5. If each vector is loaded with a single Vpr or CDC6 and a single hTERT promoter, then am I correct in understanding that theoretically these will react with a single cancer cell? I am looking forward to seeing you this Sunday. Best regards. Yoshihisa Abo,MD. Kitaaoyama D clinic, Tokyo, Japan. 1). Yes, in our TATK-based lentivirus backbone, it contains Vpr coding sequences and driven by its LTR promoter sequences; 2). The lentivital vector used in 2006 clinical trail contained Vpr sequences; 3). I do have lentiviral vector expressing Vpr protein and the expression of Vpr is controlled by hTERT promoter sequences; 4). I haven’t made any calculation of how many recombinant RNA in each milliliter of AJS2001. I do have that of the lentiviral protein p24: 1.5 mg per ml of conditioned medium, and 450 ug/ml in 300 folds concentrated AJS2001. In Cytonacx, it is unable to calculate recombinant RNA because the nanoparticles carrying plasmid DNA instead of RNA. For most cases, it contains about 700-1,000 ug of plasmid DNA per ml of concentrated nanoparticles; 5). Theoretically, it is. However, recent researches have found that the lentiviral producing cells secret many exsomes or microvesicles carrying viral RNA or recombinant RNA. These exsomes or microvesicles can transfer the recombinant RNA and some help proteins to adjacent cells, which causes “spreading” effects when AJS2001 is used. We are current doing more examines about this matter. I would thank you for your support and help the AJS2001 clinical trail. I am looking forward to seeing you in Tokyo this weekend. Very best wishes to you! Luo